
ADMET Profiling and Stability Assessment of FDA-Approved Type 2 Diabetes Drugs
- MetroTech Institute

- Jul 19, 2025
- 13 min read
Authors: Monsurat M. Lawal, Ph.D (1), Salman Siddiqi (2), Ariana Hung (3), Ganghee Lefkowitz (4)
Editor: Tugba G. Kucukkal, Ph.D
Author Affiliations: (1) MetroTech Institute, (2) Willowbrook High School, (3) Diamond Bar High School, and (4) Westlake High School, Westlake Village, CA
Abstract
Type 2 diabetes management requires a comprehensive understanding of therapeutic measures, including pharmacokinetic and pharmacodynamic properties of antidiabetic drugs. This study analyzes the ADMET-S (Absorption, Distribution, Metabolism, Excretion, Toxicity, and Stability) profiles of FDA-approved type 2 diabetes medications using computational methods. This study elucidates the relationships between molecular structure, physicochemical properties, and ADMET profiles to inform rational drug design strategies for improved therapeutic agents. We employed tools such as ADMETlab, SwissADME, and quantum mechanics-based calculations (e.g., HOMO-LUMO energy gaps) with ORCA to evaluate these drugs' stability, toxicity, and metabolic interactions. Key findings include insights into the structural determinants influencing drug stability, toxicophoric substructures, and metabolic liabilities. These results advance computational drug discovery and provide a foundation for designing next-generation antidiabetic molecules with enhanced ADMET-S profiles.
Introduction
Diabetes is a chronic metabolic disorder characterized by the body's inability to regulate blood sugar levels effectively [1]. Type 2 diabetes, the most prevalent form, arises from insulin resistance or insufficient insulin production. Prolonged hyperglycemia can lead to severe complications, including cardiovascular disease, neuropathy, and renal failure [2]. Effective management of type 2 diabetes typically involves lifestyle modifications, such as dietary changes and increased physical activity, supplemented by pharmacological interventions (Figure 1).

Several drug classes are employed in treating type 2 diabetes, each targeting distinct mechanisms to control blood glucose levels. Metformin, often the first-line therapy, enhances insulin sensitivity and reduces hepatic glucose production [3]. Sulfonylureas (e.g., Glyburide, Glipizide) and meglitinides (e.g., Repaglinide, Nateglinide) stimulate insulin secretion from the pancreas, albeit with differing durations of action. Thiazolidinediones (e.g., Pioglitazone, Rosiglitazone) improve insulin sensitivity in peripheral tissues, while DPP-4 inhibitors (e.g., Sitagliptin, Saxagliptin) enhance the body's natural glucose-lowering hormones. SGLT2 inhibitors (e.g., Canagliflozin, Empagliflozin) promote glucose excretion via the kidneys, and alpha-glucosidase inhibitors (e.g., Acarbose, Miglitol) slow carbohydrate digestion in the intestines. Despite their efficacy, these drugs exhibit variability in their pharmacokinetic and pharmacodynamic properties, necessitating a deeper understanding of their ADMET-S profiles to optimize therapeutic outcomes.
Herein, we analyze the ADMET-S properties of FDA-approved type 2 diabetes drugs using computational approaches. This analysis provides critical insights into how these compounds interact with biological systems at the molecular level, informing strategies for designing improved antidiabetic molecules. The primary objective of this study is to comprehensively evaluate the ADMET-S properties of FDA-approved type 2 diabetes drugs to understand their interactions with the human body. Objectives include analyzing ADMET properties, predicting molecular stability, identifying trends and relationships, and predicting structural determinants.
Methodology
Our methodology involved a systematic, multistep approach to evaluate the ADMET-S properties of FDA-approved type 2 diabetes drugs. The first step is drug identification and structure retrieval from the DrugBank [4]. The Simplified Molecular Input Line Entry System (SMILES) representations of these drugs were retrieved for computational analysis, and the three-dimensional (3D) structures of the drugs were obtained from PubChem [5] for further processing. The second step is ADMET Screening with ADMETlab [6] and SwissADME [7] for absorption, distribution, metabolism, excretion, and toxicity properties. The third step is molecular stability calculation, which requires 3D structure modeling in Avogadro [8] software for quantum mechanics calculations for Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energy values computation within ORCA [9]. The final step is data interpretation and result analysis.
Results and Discussion
3.1 Physicochemical Properties
Our analysis of the physicochemical properties of FDA-approved type 2 diabetes drugs revealed significant variations in their molecular characteristics. Parameters such as molecular weight (MW), van der Waals (vdW) volume, total polar surface area (TPSA), melting point (MP), boiling point (BP), and molar refractivity (MR) were examined (Figure 2a) to understand their influence on drug behavior in biological systems. Among the drugs analyzed, Acarbose exhibited the highest values across all five metrics, underscoring its substantial bulk properties. Other compounds with higher MWs and volumes are Bromocriptine, Atorvastatin, and Gliquidone. Metformin emerged as a standout compound with the lowest molecular weight, van der Waals volume, and molar refractivity (Figure 2b). Its simplicity in structure correlates with more predictable pharmacokinetic behavior, facilitating easier management and accurate dosing. These characteristics underscore the advantages of structurally simpler drugs in pharmacological reliability and patient compliance. These properties are critical determinants of a drug's absorption, distribution, and elimination. For instance, higher molecular weight or larger van der Waals volume can hinder a drug's ability to traverse cell membranes, potentially limiting its bioavailability. This outcome might be why Acarbose or any other alpha-glucosidase inhibitor is no longer recommended [10]. Similarly, elevated TPSA values may impede permeability through barriers, such as the blood-brain barrier (BBB) and intestinal epithelium.
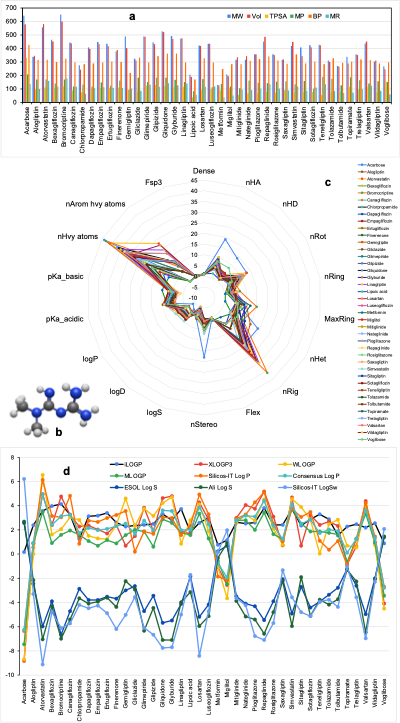
We created a radar chart to visualize the physicochemical properties of the 40 antidiabetic drug molecules (Figure 2c). These charts enable the simultaneous comparison of multiple properties, providing insights into the structural features of each drug. For example, lipophilicity and solubility (Figure 2d) are fundamental properties that govern a drug's absorption and distribution within the body. Lipophilicity, measured by LogP values, reflects a drug's ability to dissolve in lipid environments, which is crucial for traversing cell membranes [7]. Optimal lipophilicity ensures efficient absorption and distribution, enhancing the drug's therapeutic efficacy. However, excessively high lipophilicity can lead to poor solubility and increased toxicity. Solubility directly impacts a drug's bioavailability. Poorly soluble drugs often exhibit limited absorption, diminishing their effectiveness. Our analysis shows that known compounds with proven clinical efficacy demonstrate suboptimal solubility or lipophilicity. This discrepancy highlights the need for further optimization to improve their pharmacokinetic profiles.
3.2 Drug Rules and Medicinal Chemistry
To evaluate the drug-likeness of FDA-approved type 2 diabetes medications, we assessed their compliance with established medicinal chemistry rules. These rules serve as guidelines to predict a compound's pharmacokinetic behavior, bioavailability, and potential efficacy. Pioglitazone and Sitagliptin violate Pfizer's medicinal chemistry rules. Such violations often indicate structural features that may hinder drug absorption, distribution, or metabolism. Lipinski's Rule of Five [11] is widely used and states that an ideal drug candidate should have no more than five hydrogen bond donors, ten hydrogen bond acceptors, a molecular weight below 500 Da, and a logP value less than 5 to ensure optimal lipophilicity. Drugs like Acarbose and Atorvastatin contravene these criteria, suggesting potential challenges in bioavailability or stability. Beyond Lipinski's Rule, several drugs showed deviations across additional parameters, further impacting their drug-likeness. 22 of the 40 compounds violate the GSK rule, Acarbose, Atorvastatin, Bromocriptine, Gliquidone, and Metformin failed the GoldenTriangle rule, 13 of the 40 drug molecules, including Acarbose and Atorvastatin violate the Ghose rule. We also recorded seven drugs, including Acarbose and Atorvastatin, violating the Veber drug rule. Acarbose, Atorvastatin, Glimepiride, Glipizide, and Voglibose did not align with the Egan theory, seven drugs, including Gemigliptin, violate the Muegge rule, 9 of the 40 compounds have Brenk violation, and only 10 over 40 drugs, including Alogliptin, are potential lead candidates. Violations of these rules indicate reduced efficacy, increased toxicity, or poor pharmacokinetic profiles. Such findings highlight the importance of adhering to medicinal chemistry principles during drug design to minimize adverse effects and enhance therapeutic outcomes.
3.3 Absorption and Distribution
The BOILED-Egg model [12] was employed to predict the absorption and distribution properties of the antidiabetic drugs. This model provides a graphical representation of a topological polar surface area (TPSA) versus logP (Figure 3), offering insights into critical pharmacokinetic attributes. The yellow region of the BOILED-Egg plot indicates BBB permeability, while the white/gray region represents human intestinal absorption (HIA). The BBB is a selective barrier that protects the central nervous system (CNS) from harmful substances while permitting the passage of essential molecules. Drugs targeting CNS disorders must possess specific physicochemical attributes, including low molecular weight, moderate lipophilicity, and limited polar surface area, to effectively penetrate the BBB. Our findings indicate that drugs with higher molecular weights and polar surface areas are less likely to cross this barrier. The intestinal epithelium plays a pivotal role in regulating the absorption of orally administered drugs. Factors such as solubility, stability, and permeability significantly impact oral bioavailability. High molecular weight and sizeable polar surface area can restrict absorption through the intestinal barrier, reducing the drug's effectiveness.
Additionally, the model identifies whether a drug interacts with P-glycoprotein (PGP), a key transporter involved in drug efflux. Nateglinide and Pioglitazone demonstrated the highest scores in terms of absorption and distribution, suggesting favorable pharmacokinetic profiles. These findings underscore their ability to reach target tissues and exert therapeutic effects. Understanding absorption and distribution is crucial for predicting how well a drug can penetrate biological barriers and achieve optimal concentrations at its target site. Poor absorption or limited distribution can significantly reduce a drug's effectiveness, emphasizing the need for careful optimization of these properties during drug development.

Our analysis also revealed the unique pharmacokinetic properties of certain antidiabetic drugs, providing valuable insights into their behavior within the body. Drugs, such as Mitiglinide, Nateglinide, and Sitagliptin, exhibited the ability to permeate the BBB. Mitiglinide and Nateglinide possess amide and carboxylic functional groups and mimic peptide derivatives, facilitating their passage through the BBB. This characteristic could be advantageous for drugs targeting central nervous system-related complications of diabetes. Voglibose and Acarbose deviated from the BOILED-Egg model due to their highly decorated structures, featuring multiple hydroxyl units. These structural features likely contribute to their poor absorption and distribution profiles, limiting their pulled effectiveness. Miglitol and Atorvastatin demonstrated suboptimal absorption and distribution, raising concerns about their therapeutic efficacy. These findings suggest the need for reformulation strategies or combination therapies to enhance their performance. Most molecules have bioavailability scores of 0.55 and 0.56, except Acarbose, with a lower value of 0.17, and Nateglinide and Pioglitazone, with the highest score of 0.85.
3.4 Metabolism of Type 2 Diabetes Drugs
Understanding the metabolic behavior of antidiabetic drugs is essential for predicting their elimination rates, potential drug-drug interactions, and overall safety profiles. CYP-mediated metabolism could lead to the formation of toxic metabolites and the occurrence of undesirable drug-drug interactions [13]. Our analysis focuses on the probability of these drugs acting as substrates or inhibitors of Cytochrome P450 (CYP) enzymes, which play a central role in Phase I metabolism (oxidation, reduction, and hydrolysis). Additionally, we evaluated their stability in human liver microsomes (HLM). Drugs such as Sotagliflozin and Glyburide exhibited a high predicted affinity for CYP enzymes, indicating significant metabolic interactions (Figure 4). These interactions can influence the rate of drug elimination and increase the risk of adverse effects due to competition with other medications metabolized by the same enzymes. For instance, co-administering drugs with overlapping metabolic pathways may lead to altered pharmacokinetics, necessitating careful monitoring or dosage adjustments. High-affinity compounds often act as substrates or inhibitors of CYP enzymes, impacting their metabolic stability and potential toxicity. This competition for enzyme binding sites can result in reduced clearance rates, prolonged drug exposure, and increased risk of side effects. Understanding these interactions is crucial for optimizing therapeutic regimens and minimizing the likelihood of adverse events.
We also analyze the structural features of drugs with high CYP affinity to further elucidate the relationship between molecular structure and metabolic behavior. Unique functional groups identified as key contributors to high CYP affinity include halides (e.g., chlorine), sulfides, sulfonamides, amides, and ethers (Figure 4b). These moieties enhance the likelihood of a drug interacting with CYP enzymes, potentially affecting its metabolic stability and toxicity profile. Drug molecules with high CYP affinity may require tailored dosing strategies or close monitoring to mitigate the risk of adverse effects. For example, dose adjustments may be necessary when these drugs are co-administered with other medications sharing similar metabolic pathways. This analysis provides valuable insights into the structural determinants of CYP interactions, guiding future efforts to design drugs with improved metabolic properties.
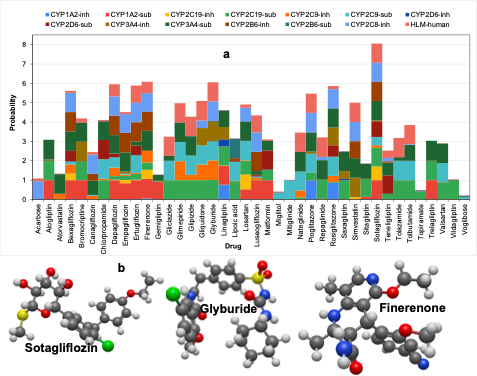
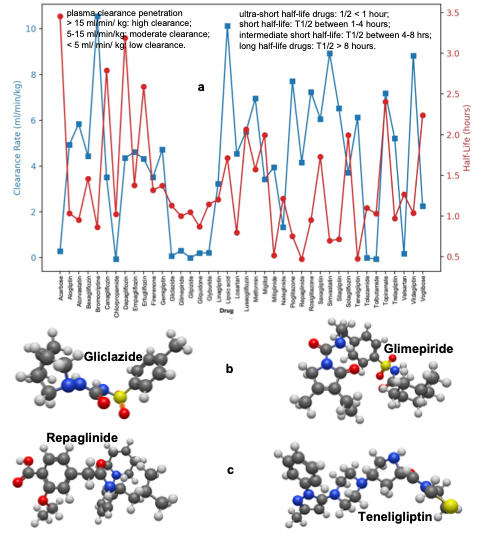
3.5 Excretion of Antidiabetic Drugs
Excretion metrics, including half-life and plasma clearance (Figure 5), are critical for understanding drug elimination and optimizing dosage regimens. Our analysis categorizes antidiabetic drugs based on their half-life and plasma clearance rates. Ultra-short half-life (<1 hour) drugs like Repaglinide and Teneligliptin are rapidly eliminated from the body, necessitating frequent dosing to maintain adequate concentrations. Short half-life (1–4 hours) drugs exhibit moderate elimination rates, requiring careful consideration of dosing intervals. Intermediate half-life (4–8 hours) drugs maintain a balance between sustained therapeutic effects and manageable dosing frequency. Long half-life (>8 hours) drugs with extended half-lives provide prolonged therapeutic benefits but carry an increased risk of accumulation and toxicity if not carefully monitored.
High clearance (>15 ml/min/kg) indicates that drugs are eliminated quickly, reducing the risk of accumulation but requiring more frequent dosing. Moderate clearance (5–15 ml/min/kg) drugs exhibit intermediate elimination rates, depicting a compromise between efficacy and safety. Low clearance (<5 ml/min/kg) drugs like Gliclazide and Glimepiride have low clearance rates, leading to prolonged systemic exposure. While this can help maintain therapeutic levels, it also increases the risk of toxicity, underscoring the need for careful monitoring.
3.6 Toxic Properties of Antidiabetic Drugs
Toxicity assessment is a critical component of drug safety evaluation. We identify toxicophoric substructures, toxicity rules, and several toxicity metrics within the molecular frameworks of FDA-approved type 2 diabetes drugs. Voglibose, Miglitol, and Luseogliflozin exhibited no toxic substructures under eight toxicity rules available from ADMETlab [6]. These compounds are characterized by at least four hydroxyl groups, which could contribute to their favorable safety profiles. In contrast, Acarbose displayed significant toxicophoric components, raising potential safety concerns. Similarly, the pyrrole ring in Bromocriptine and the chirality of SGLT2 inhibitors were identified as predictably toxic features. These findings highlight the importance of early identification of toxicity properties to refine drug design and minimize adverse effects. For instance, modifying the chemical structure of Acarbose could reduce its toxicity while preserving its therapeutic efficacy. Understanding the toxicophoric substructures of existing drugs provides valuable insights for rational drug design. By targeting these structural features, future formulations can be optimized to enhance safety and mitigate risks associated with long-term use.

3.7 Molecular Stability Prediction
Molecular stability is a key determinant of a drug's shelf life and efficacy during administration. We analyze the HOMO-LUMO energy difference (band gap) of these diabetes molecules to predict their stability. The higher the energy gap, the higher the molecular stability, indicating a reduction in the likelihood of degradation. Linagliptin and Topiramate have the lowest (3.8 eV) and highest (7.2 eV) energy gap, respectively (Figure 7). Ensuring molecular stability is crucial for maintaining drug efficacy throughout its shelf life and during patient administration.

Conclusion and Recommendations
This study comprehensively analyzes the ADMET-S (absorption, distribution, metabolism, excretion, toxicity, and stability) properties of FDA-approved type 2 diabetes drugs. Key findings include significant variations in molecular weight, lipophilicity, solubility, and polar surface area, influencing drug absorption, distribution, and elimination. Violations of medicinal chemistry rules, such as Lipinski's Rule of Five, were identified, impacting bioavailability and stability. The BOILED-Egg model revealed differences in blood-brain barrier permeability and intestinal absorption, highlighting the importance of optimizing these properties for enhanced therapeutic outcomes. High CYP affinity and low plasma clearance rates were identified as factors affecting drug elimination and potential toxicity. Toxicophoric substructures and HOMO-LUMO energy gaps provided insights into drug safety and stability, which could guide future formulation improvements.
Future studies should involve coupling these molecules with target proteins and performing molecular dynamics simulations to evaluate their detailed bio-interactions and efficacy. We recommend targeting structural determinants influencing ADMET-S profiles to design next-generation antidiabetic drugs with enhanced safety, stability, and potency. Also, modifying chemical structures, particularly for compounds like Acarbose, can reduce toxicity and improve stability, ensuring better therapeutic outcomes. Finally, advanced computational tools, including artificial intelligence and machine learning, should be leveraged to refine ADMET predictions and streamline drug discovery pipelines.
References
Holt, R.I., Cockram, C.S., Ma, R.C., Luk, A.O.: Diabetes and infection: review of the epidemiology, mechanisms and principles of treatment. Diabetologia. 1–13 (2024)
Al-Faqeeh, L.A.S.: Diabetes Mellitus: A Health Syndrome. In: Therapeutic Mushrooms for Diabetes Mellitus. pp. 1–11. Apple Academic Press (2023)
Dean, L., McEntyre, J.: Introduction to Diabetes. In: The Genetic Landscape of Diabetes
Knox, C., Wilson, M., Klinger, C.M., Franklin, M., Oler, E., Wilson, A., Pon, A., Cox, J., Chin, N.E., Strawbridge, S.A.: DrugBank 6.0: the DrugBank knowledgebase for 2024. Nucleic Acids Res. 52, D1265–D1275 (2024)
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., Li, Q., Shoemaker, B.A., Thiessen, P.A., Yu, B.: PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380 (2023)
Fu, L., Shi, S., Yi, J., Wang, N., He, Y., Wu, Z., Peng, J., Deng, Y., Wang, W., Wu, C.: ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 52, W422–W431 (2024)
Daina, A., Michielin, O., Zoete, V.: SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 1–13 (2017)
Hanwell, M.D., Curtis, D.E., Lonie, D.C., Vandermeersch, T., Zurek, E., Hutchison, G.R.: Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics. 4, 1–17 (2012)
Neese, F.: Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 12, e1606 (2022)
Standards of Care in Diabetes
Lipinski, C.A.: Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 101, 34–41 (2016)
Daina, A., Zoete, V.: A boiled‐egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 11, 1117–1121 (2016)
Oladipo, S.D., Zamisa, S.J., Badeji, A.A., Ejalonibu, M.A., Adeleke, A.A., Lawal, I.A., Henni, A., Lawal, M.M.: Ni2+ and Cu2+ complexes of N-(2, 6-dichlorophenyl)-N-mesityl formamidine dithiocarbamate structural and functional properties as CYP3A4 potential substrates. Sci. Rep. 13, 13414 (2023)



Comments